Scoperta di una mutazione genetica rara e il suo impatto sulla neurodegenerazione
Recenti ricerche hanno rivelato una mutazione genetica estremamente rara, responsabile di una forma di neurodegenerazione nei bambini. Questo studio, condotto da un team di scienziati del Helmholtz Munich, ha messo in luce un nuovo meccanismo attraverso il quale le cellule cerebrali possono morire. I risultati suggeriscono che meccanismi simili di morte cellulare potrebbero essere implicati in altre patologie neurologiche, come la ferroptosi, il Parkinson e la malattia di Huntington. La comprensione di questi meccanismi è fondamentale per sviluppare nuove strategie terapeutiche e migliorare la qualità della vita dei pazienti affetti da queste malattie.
Il legame tra mutazioni genetiche e morte cellulare nei neuroni
La ricerca ha evidenziato come specifiche mutazioni all’interno di un singolo gene possano innescare un processo di infiammazione progressiva e conseguente morte cellulare nei neuroni di modelli murini. Sorprendentemente, anche le cellule cerebrali umane, coltivate in laboratorio e derivate da fibroblasti di pazienti affetti dalla stessa mutazione, hanno mostrato un tasso di mortalità neuronale simile a quello osservato nei topi. Questo suggerisce che le scoperte fatte in modelli animali possano avere rilevanza clinica per la comprensione delle malattie neurodegenerative negli esseri umani.
Ferroptosi: un meccanismo di morte cellulare da esplorare
Il fenomeno di morte cellulare programmata identificato in questo studio è noto come ferroptosi. Questo processo è caratterizzato dall’accumulo di ferro e dal danno ossidativo alle membrane cellulari. I ricercatori hanno osservato che il meccanismo di ferroptosi potrebbe avere analogie con i processi di morte cellulare che si verificano nella demenza. Questa ipotesi è supportata da un’analisi approfondita delle proteine espresse dai neuroni. Recenti studi hanno, infatti, suggerito un legame tra ferroptosi e malattia di Alzheimer, aprendo nuove strade per la ricerca terapeutica.
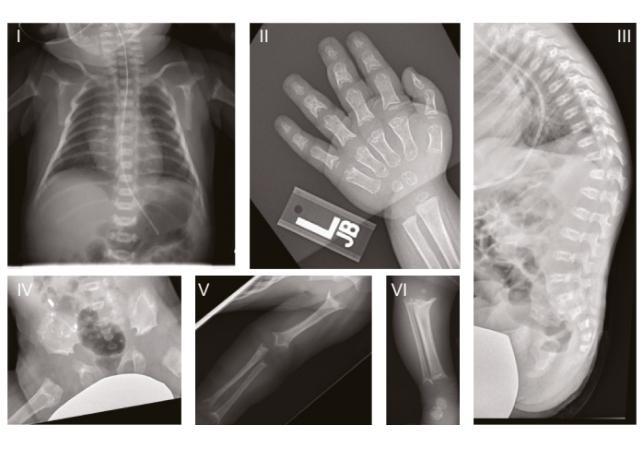
Displasia spondilometafisaria di tipo Sedaghatian: un disturbo raro
Il disturbo genetico in questione, noto come displasia spondilometafisaria di tipo Sedaghatian (SSMD), è caratterizzato da gravi anomalie cerebrali e scheletriche. Descritto per la prima volta nel 1980, questo disturbo è estremamente raro, con solo pochi casi documentati, molti dei quali riguardano bambini che purtroppo non sopravvivono a lungo. La rarità di questo disturbo rende fondamentale la ricerca per comprendere meglio le sue cause e le possibili terapie.
Il ruolo cruciale dell’enzima GPX4 nella neuroprotezione
Negli ultimi anni, il sequenziamento genomico ha rivelato che la SSMD è associata a mutazioni nel gene che codifica per l’enzima GPX4, considerato un “guardiano” della ferroptosi per la sua capacità di proteggere le membrane cellulari dai danni ossidativi. Sebbene le mutazioni in questo gene non conducano necessariamente a una demenza a esordio precoce, la ricerca attuale su modelli murini e organoidi cerebrali coltivati in laboratorio ha messo in evidenza il ruolo cruciale del GPX4 nella protezione neuronale e come la sua disfunzione possa portare alla morte cellulare. Comprendere il funzionamento di questo enzima è essenziale per sviluppare interventi terapeutici mirati.
Studi sui pazienti affetti da SSMD e implicazioni future
Lo studio ha esaminato tre bambini statunitensi affetti da SSMD, i quali presentavano vari gradi di atrofia cerebrale e mutazioni nella stessa regione funzionale del gene GPX4. Questi dati sono stati utilizzati per ulteriori esperimenti su topi e cellule cerebrali coltivate in laboratorio, ottenute da fibroblasti di un paziente con SSMD. La ricerca continua a esplorare le connessioni tra mutazioni genetiche e neurodegenerazione, con l’obiettivo di identificare potenziali trattamenti per migliorare la vita dei pazienti.
La metafora dell’enzima GPX4 e la sua funzione protettiva
Marcus Conrad, biologo cellulare e direttore dell’Istituto di Metabolismo e Morte Cellulare di Helmholtz Munich, ha paragonato l’enzima GPX4 a una tavola da surf. Con la sua pinna immersa nella membrana cellulare, l’enzima scivola lungo la superficie interna, disintossicando rapidamente i perossidi lipidici. Tuttavia, in presenza della mutazione specifica del GPX4, la pinna è assente, il che significa che l’enzima non è ancorato alla membrana e non può svolgere la sua funzione protettiva nei neuroni. Questa scoperta sottolinea l’importanza di comprendere le interazioni molecolari per affrontare le malattie neurodegenerative.
Ferroptosi come forza trainante nella morte neuronale
I neuroni coltivati in laboratorio, derivati da cellule staminali di pazienti con SSMD, si sono rivelati particolarmente suscettibili alla ferroptosi. L’interruzione di questo processo nei modelli murini e nelle cellule coltivate, mediante l’uso di un composto chimico, ha mostrato di rallentare la morte neuronale. “I nostri dati suggeriscono che la ferroptosi potrebbe essere una forza trainante nella morte neuronale, piuttosto che un semplice effetto collaterale”, ha affermato Svenja Lorenz, biologa cellulare di Helmholtz Munich. Fino ad ora, la ricerca sulla demenza si è concentrata prevalentemente sui depositi proteici nel cervello, noti come placche di amiloide β. Tuttavia, ora si sta ponendo maggiore attenzione al danno alle membrane cellulari, che potrebbe essere il fattore scatenante di questa degenerazione.
Demenza infantile e la necessità di ulteriori ricerche
Sebbene la demenza sia comunemente associata all’età avanzata, in alcuni casi tragici, il declino cognitivo legato a problemi di memoria può manifestarsi in età molto precoce. La demenza infantile è una condizione cerebrale rara che porta a perdita di memoria e confusione, e studi genomici l’hanno collegata a oltre 100 disturbi rari presenti alla nascita. L’analisi di tali casi fornisce agli scienziati preziose informazioni su come avvenga la neurodegenerazione e su quali possano essere le possibili strategie di intervento. “Ci sono voluti quasi 14 anni per collegare un piccolo elemento strutturale di un singolo enzima a una grave malattia umana”, ha commentato Conrad. “Progetti come questo dimostrano chiaramente l’importanza di investimenti a lungo termine nella ricerca di base e la necessità di team multidisciplinari internazionali per comprendere appieno malattie complesse come la demenza e altre condizioni neurodegenerative”. I risultati di questo studio sono stati pubblicati sulla rivista scientifica Cell.